
| Home | Syllabus | Schedule | Lecture Notes | Extras | Glossary |
| Home | Syllabus | Schedule | Lecture Notes | Extras | Glossary |
We began with the announcement from Andy Nguyen at CAPS that there are Supplemental Instruction sessions at the following times:
We continue our exploration of the biochemistry of heredity, beginning with a review of the properties of DNA polymerase I (the Kornberg polymerase). Last time we saw that DNA polymerase I has a 5' to 3' DNA polymerase activity, responsible for strand extension on a primed template. It also has a 3' to 5' exonuclease activity that is used for proofreading; if an incorrect base is added to the growing strand, the polymerase backs up and removes it.
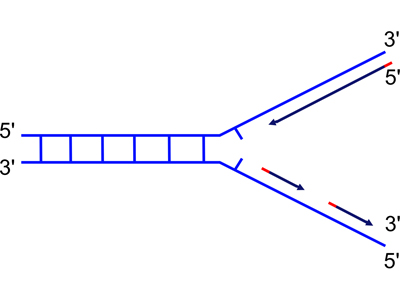
There is a third enzymatic activity of DNA polymerase I: a 5' to 3' exonuclease. We discussed why DNA polymerase might need a 5' to 3' exonuclease for a while, until someone hit upon the idea of completing the synthesis of the discontinuous strand. Consider the problem of replicating both strands at the replication fork, as illustrated below.
 |
 |
| Because DNA can only be synthesized from the 5' to 3' direction, the two newly-synthesized strands are synthesized differently. They are called the "continuous" (on the top in the figure) and "discontinuous" (on the bottom in the figure) strand, and also the "leading" (top) and "lagging" (bottom) strand. | As the replication fork moves down the helix, you can see the leading strand synthesized continuously, while the lagging strand must initiate new sites of synthesis. |
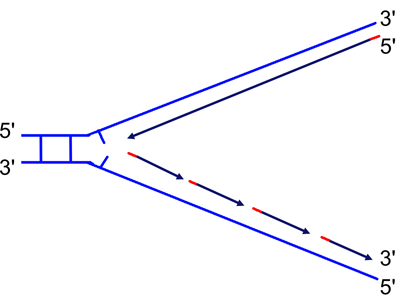
In order to complete the synthesis of the discontinuous strand, DNA polymerase must elongate each Okazaki fragment until it reaches the 5' end of the next Okazaki fragment. This will leave a nick in place, but also another problem: the 3' end of the completed Okazaki fragment is DNA, but the 5' end of the next Okazaki fragment is RNA synthesized by primase. In order to completely replicate the DNA molecule, the RNA must be removed. This is the function of the 5' to 3' exonuclease, as shown below.
 |
We begin with a mostly-replicated strand with a one-base gap before we encounter an RNA primer shown in red (note the U). We can imagine either that DNA polymerase is about to add the last base of the Okazaki fragment, or that a single base has been removed due to mismatch repair. |
 |
The 5' to 3' exonuclease activity removes the RNA primer as the 5' to 3' DNA polymerase activity synthesizes the new DNA strand. |
 |
Here the RNA primer has been completely removed, leaving only a nick to be sealed by DNA ligase. Compare the second to the third drawing to see how DNA polymerase is capable of moving ("translating") a nick from the 5' to 3' direction. This is called "nick translation." |
The simultaneous consideration of genetics and biochemistry in which we are engaged makes us ask what sort of difficult question a geneticist might ask a biochemist who had just presented all of this information on DNA polymerase. There were some fairly good questions proposed, none as hard as this one: how do you know that DNA polymerase I is actually the enzyme that replicates the bacterial chromosome in E. coli?
The question was addressed using a brute-force screen for mutations affecting the activity of DNA polymerase. Colonies of E. coli, derived from individual mutagenized cells, were grown in culture to obtain cells derived from single colonies. Each culture was assayed for the Kornberg DNA polymerase. Imagine the surprise of the scientific community when a strain of E. coli lacking functional DNA polymerase I was obtained. This demonstrates that DNA polymerase I actually only plays a minor role in chromosomal DNA replication; its principle function is mismatch repair.
In our concept map for DNA to Trait, we have three ideas: 1) genes are made of DNA (as demonstrated by the Hershey-Chase experiment), 2) genes encode proteins (to be developed here), and 3) DNA is code (to be explored at the end of this talk).
The idea that genes encode proteins has its origins in the work of Archibald Garrod, whose investigations of alkaptonuria began in 1901 just after the rediscovery of Mendel. He published a monograph discussing four inborn errors of metabolism in 1908. One of the inborn errors is albinism, which we now know to be a defect in tyrosine hydroxylase, resulting in the inability to synthesize melanin. Garrod's monograph doesn't contain much information on albinism, and we have already spent a fair amount of time on the subject.
Garrod's most detailed work was on alkaptonuria, a peculiar but mostly harmless inborn error of metabolism. Alkaptonuria is an autosomal recessive disorder. The urine of people with alkaptonuria turns black upon exposure to air. The black substance is often present as spots in the white of the eye, and is very evident in cartilage, which is often as black as coal. In people with pale skin, you can see the black cartilage as a bluish tinge in their ears and nose.
Garrod and his colleagues investigated the source of the black substance in the urine, discovered to be homogentisic acid, a compound with a six-member carbon ring. A heavy protein meal causes more homogentisate in the urine of alkaptonurics. There are three amino acids with six-member rings in their side groups: phenylalanine, tyrosine, and tryptophan. In tryptophan the six member ring is joined to a five-member ring containing nitrogen, collectively called an indole ring. This suggested that particular amino acids might be the source of homogentisate in alkaptonurics.
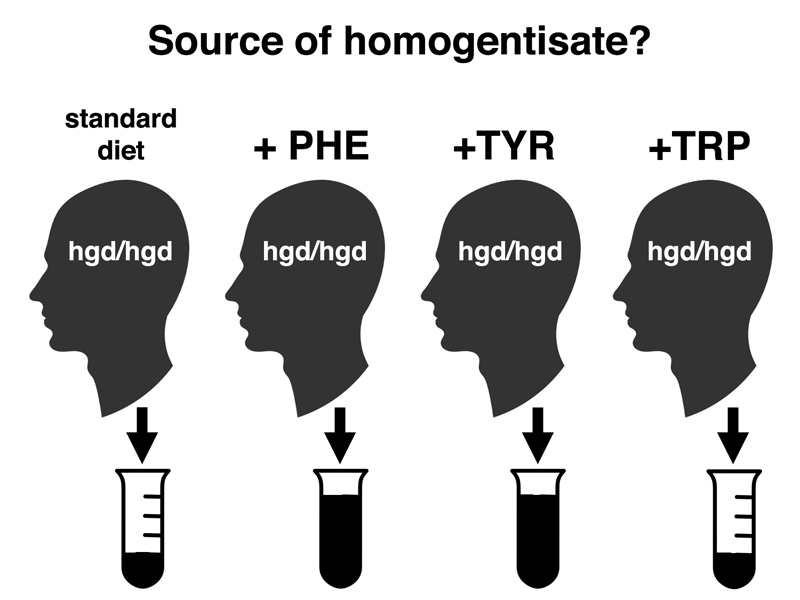
In the experiments outlined below, people with alkaptonuria (hgd/hgd) were fed a standard diet, or a standard diet supplemented with phenylalanine (PHE), tyrosine (TYR), or tryptophan (TRP). As shown in the cartoon, supplements of phenylalanine or tyrosine produce a large increase in the quantity of homogentisate in the urine, while tryptophan has no effect. This suggests that alkcaptonurics metabolize phenylalanine and tyrosine to homogentisate, but it doesn't tell us whether normal people use that pathway or whether this is just a curiosity seen in alkaptonurics.
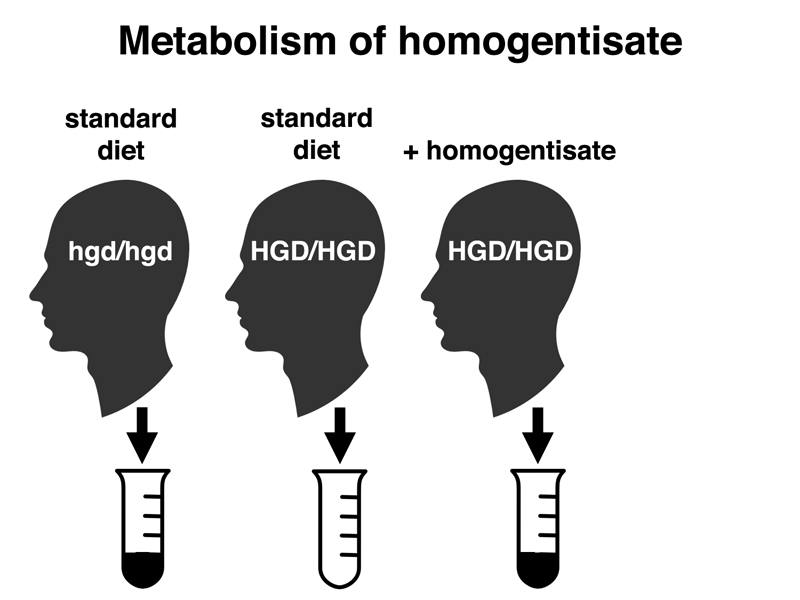
In the experiments shown in the cartoon below, we see that normal people (HGD/HGD) do not produce homogentisate in their urine. However, if they are given a large amount (several grams) of homogentisate, they will produce black urine transiently. Smaller doses of homogentisate have no effect, showing that normal people are capable of metabolizing homogentisate, and that alkaptonurics lack this ability.
We can conclude the following from our study of alkaptonuria:
In 1941, Beadle and Tatum provided convincing evidence that genes controlled the synthesis of enzymes, with each gene specifying a different enzyme. Beadle had started to work on the genetic control of biochemical pathways in Drosophila, working on the synthesis of eye pigments, but switched to a simpler genetic system when he began to work with Tatum.
Beadle and Tatum worked with the common bread mold Neurospora. The life cycle of Neurospora is shown below.
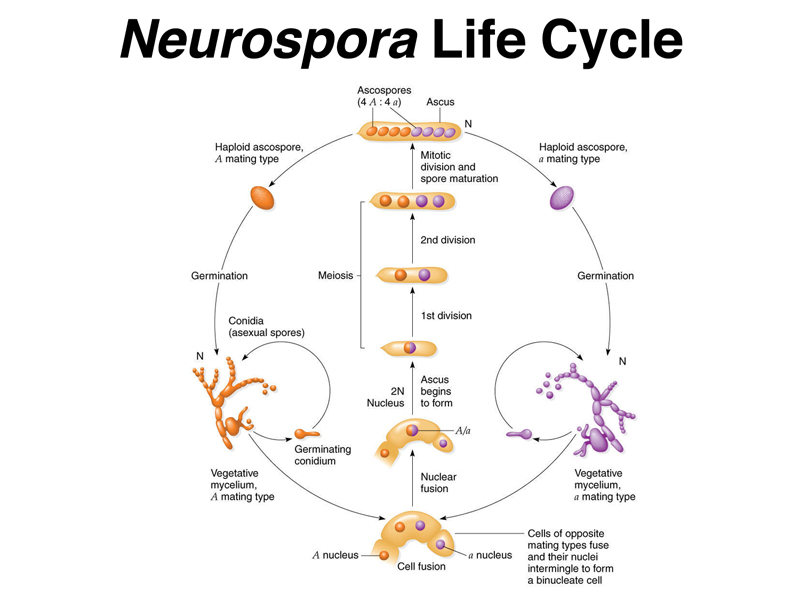
As shown in the figure, the haploid phase dominates the life cycle of Neurospora. Haploid vegetative mycelium grows and makes conidia (asexual spores). When cells of opposite mating types meet, they fuse to create cells with two different types of nuclei, which then fuse to make a diploid nucleus that immediately enters meiosis. The four meiotic products undergo a single postmeiotic mitotic division to make eight ascospores. The eight ascospores are enclosed in a structure called the ascus. The ascus can be dissected to remove the spores individually to separate cultures.
The figure below shows the experimental design that Beadle and Tatum used to obtain auxotrophs of Neurospora.
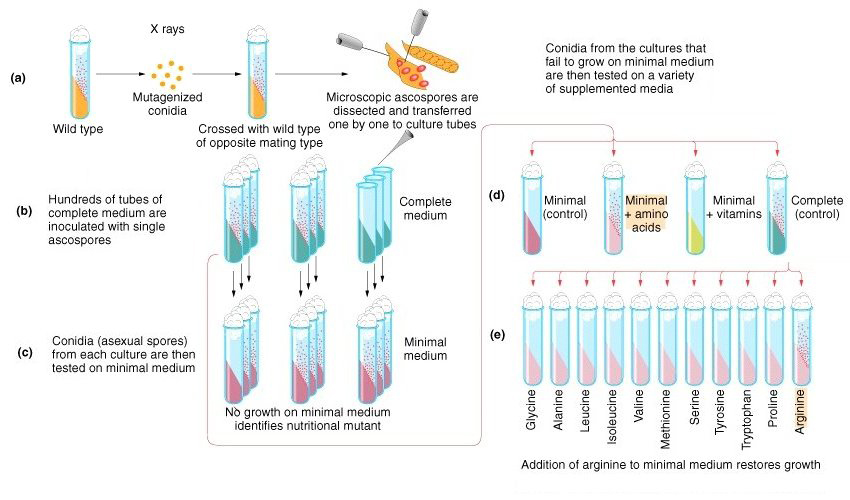
As shown in the figure, Beadle and Tatum irradiated conidia with X rays to produce mutations. They crossed the mutagenized conidia with an unirradiated wild type strain of the opposite mating type. They cultured individual ascospores separately on complete medium (medium containing a very rich mix of nutrients), collecting hundreds of cultures.
They tested the ability of each culture to grow on synthetic minimal medium (medium containing a carbon source, an inorganic nitrogen source, and simple salts). Cultures that could not grow on minimal medium were tested for their ability to grow on minimal medium plus supplements (a mix of amino acids or a mix of vitamins) to identify the class of compounds that was needed, then on minimal medium plus specific compounds to identify the exact compound required. In the figure, a strain requiring amino acids is ultimately discovered to be an arginine auxotroph.
The arginine biosynthetic pathway is shown below.
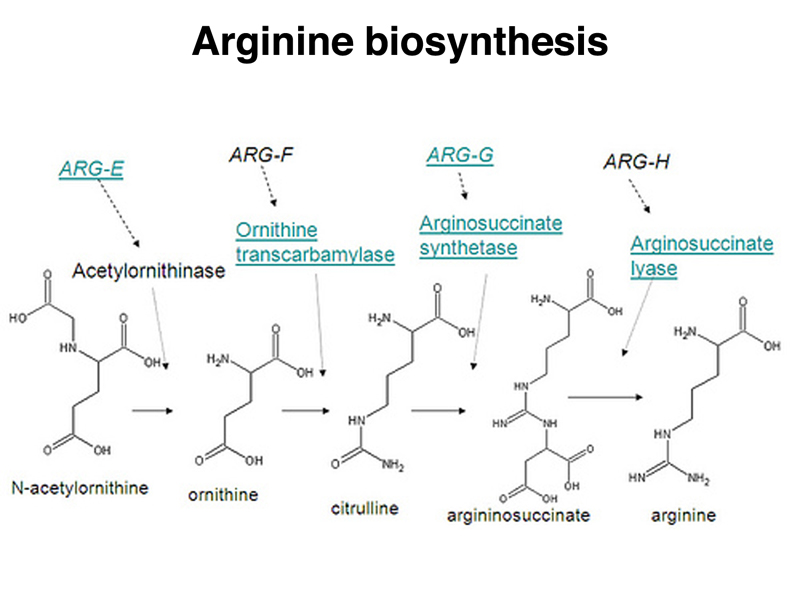
Different arginine auxotrophs differ in their ability to grow on a medium supplemented with different intermediates in the biosynthesis of arginine. There are four kinds of arginine auxotrophs, as shown in the table below.
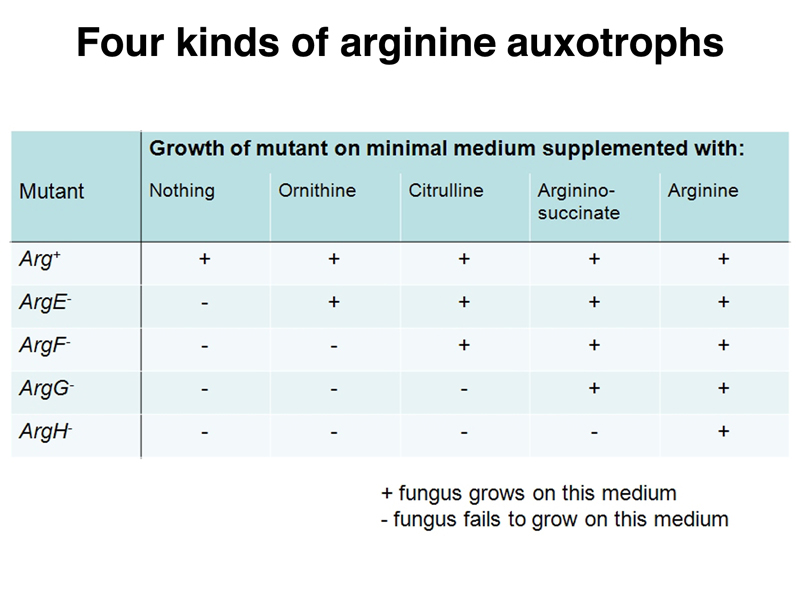
Wild-type Neurospora can grow on minimal medium without supplements, as well as on minimal medium containing any of the intermediates in arginine biosynthesis. Arginine auxotrophs can be classified by their ability to grow on minimal medium supplemented with specific intermdiates in arginine biosynthesis. Each mutant strain has a specific enzymatic defect. Arginine auxotrophs that can only grow on minimal medium supplemented with arginine have a defect in the last step of the pathway, the conversion of argininosuccinate to arginine. Arginine auxotrophs that can grow on minimal medium with any of the supplements have a defect early in the pathway, in the synthesis of ornithine.
It is possible to use genetic techniques (which we have not presented) to show that all of the independently-derived auxotrophs of one class represent independent mutations of the same gene. Therefore, individual genes control the synthesis of specific enzymes. By extension, the function of genes is to direct the synthesis of proteins. In 1941, the world was ready to hear this message, and Beadle and Tatum's "One Gene, One Enzyme" hypothesis had a big impact on the field of genetics.
Humans and other mammals are auxotrophs for a group of amino acids called "essential" amino acids. We call them essential because we need them in our diet. The essential amino acids tend to have more complex structures and require more energy to synthesize. In the course of getting enough calories to survive, we generally get more than enough amino acids to meet our needs, and end up catabolizing amino acids for energy. You can read more about essential amino acids at Wikipedia.
Humans have the biosynthetic pathway for arginine, but we use it to dispose of excess nitrogen derived from using amino acids as an energy source. The metabolic cycle that accepts ammonia derived from the deamination of amino acids and gets rid of it as urea is called the urea cycle, shown below. Urea is synthesized as part of arginine, and is released when arginine is converted to ornithine by arginase.
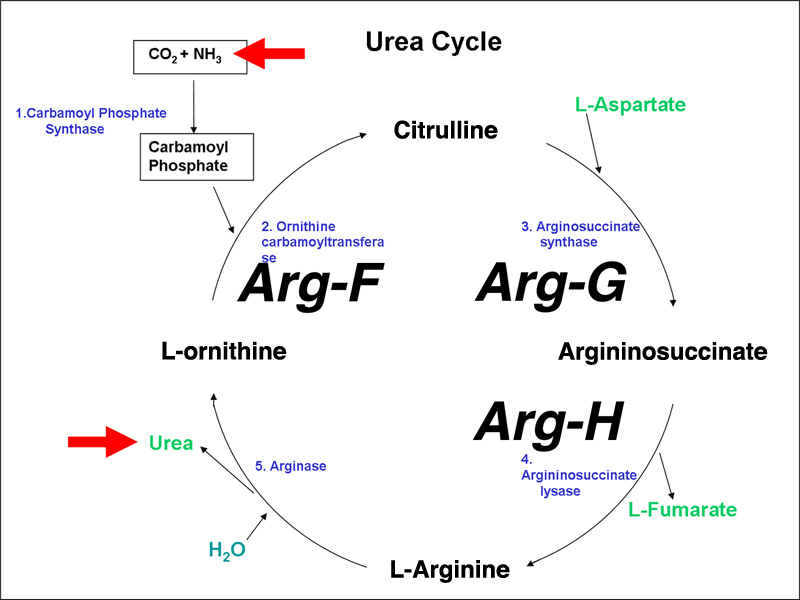
I have labeled three enzymes with the gene names from Neurospora to show that humans have the same enzymes.
Inborn errors of the urea cycle are known in humans. We do not initially recognize these as arginine auxotrophy (arginine is a nonessential amino acid in normal humans), but rather as disorders in the disposal of waste nitrogen. We will use ornithine transcarbamylase deficiency as an example. Ornithine transcarbamylase (aka ornithene carbamoyltransferase) is encoded by the OTC gene, which is sex-linked. People with mutations in this gene lack the ability to convert ornithine to citrulline, the reaction that imports ammonia into the urea cycle.
The consequence of this enzyme defect is hyperammonemia, the presence of high concentrations of ammonia in the blood. Ammonia is very toxic, and hyperammonemia typically produces confusion, ataxia, paranoia, seizures, and coma. Most patients with ornithine transcarbamylase deficiency have some enzyme function, and can be successfully treated with a low protein diet supplemented with arginine. In some cases, there is enough enzyme function to allow the person to escape detection as having a urea cycle disorder until an unusual event produces hyperammonemia. Such events might include a very high protein meal, or surgery or illness that places the person in negative nitrogen balance (catabolizing a large amount of the protein in their body).
You can read more about ornithine transcarbamylase deficiency at OMIM.
Now that we know that the function of genes (which we know to be made of DNA) is to encode proteins, we can begin to ask about the nature of the genetic code. The flow of information from DNA to protein is illustrated below.
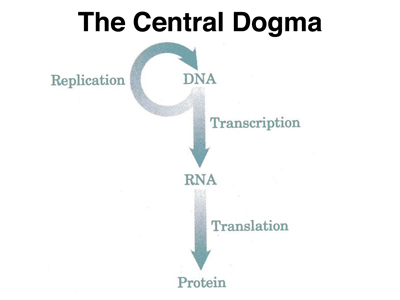
Before we get into the details of transcription and translation, we can guess that there must be some way in which a sequence of nucleic acids can correspond to a sequence of amino acids. The transcription of DNA into RNA is easy to understand at this level. We know that RNA can base pair with DNA (look at the RNA primers synthesized by primase), so making an RNA copy of a DNA sequence does not require us to understand much more than we have already discussed.
The translation of an RNA sequence to a protein sequence is more challenging. There must be a code that allows a sequence of bases to stand for an amino acid.
We know that there are twenty primary amino acids (amino acids that can be incorporated into proteins). There are many amino acids that are secondary amino acids, not incorporated into proteins. Examples of secondary amino acids are ornithine, citrulline, and argininosuccinate, whose structures are shown above in the discussion of arginine auxotrophs in Neurospora.
How many bases are required to encode the twenty primary amino acids?
If the genetic code were one base, we could only encode four different amino acids.
With a two base genetic code, we can encode 42 or 16 amino acids, which is not enough to encode the 20 primary amino acids.
With a three base genetic code, we can encode 43 or 64 amino acids, which is more than enough to encode the 20 primary amino acids. We can speculate that it is likely that the genetic code is degenerate, that is to say, that some amino acids are encoded by multiple three-base synonyms. In class discussion, it became evident that many students have heard of the genetic code, are aware that it is a three-base code, and also know that it has two kinds of punctuation: start and stop. There was also a reasonable awareness that there is no other kind of punctuation, for example commas or slashes to separate the words that stand for amino acids.
We will explore the details of the genetic code as we discuss translation in a future lecture.