
| Home | Syllabus | Schedule | Lecture Notes | Extras | Glossary |
| Home | Syllabus | Schedule | Lecture Notes | Extras | Glossary |
We began by reminding everyone that the next exam is scheduled for Tuesday, October 15. I will post a study guide on Tuesday, October 8. Fall break is scheduled for Thursday and Friday, October 10 and 11. I will be available by apppointment on those days. I will not be here on October 14 or 15. Someone else will proctor the exam. Please schedule your study time with these things in mind.
We began our discussion with a loose definition of mutation, any change in the DNA sequence of the genome of an organism that causes it to differ from what we think of as wild type.
The simplest kind of mutations to think about are "point mutations." These are substitutions of one base for another. Because there are many different chemical mutagens (agents that increase the mutation rate) that act using different mechanisms, there is a specific vocabulary used to talk about types of point mutations.
There is a kind of point mutation called a transition, in which one purine is substituted for another or one pyrimidine is substituted for another. The substitution of A for G (or vice versa) is a purine for purine substitution. The substitution of T for C (or vice versa) is a pyrimidine for pyrimidine substitution. Both types are transitions.
There is another kind of point mutation called a transversion, in which a purine is substituted for a pyrimidine. There are twice as many possible transversions as there are transitions: A can change to C or T, G can change to C or T, T can change to A or G, and C can change to A or G.
We are often concerned with the number of different codons that can result from single base changes to an ancestral codon. Taking our initial codon as AAA, we can see that there are three new codons that result from transitions, six from transversions, or nine possible changes from base substitutions overall, as shown below.
| transition | transversion | base substitution |
| GAA | CAA | CAA |
| AGA | TAA | GAA |
| AAG | ACA | TAA |
| ATA | ACA | |
| AAC | AGA | |
| AAT | ATA | |
| AAC | ||
| AAG | ||
| AAT |
When we discussed DNA replication, we saw that the error rate for the 5' to 3' DNA polymerase is about 10-3. Because DNA polymerase has a 3' to 5' exonuclease for proofreading, the error rate falls to about 10-6. There is also a mismatch repair system that functions to remove a base pair mismatch, targeting the newly synthesized strand, that gets the error rate down to around 10-9. This error rate means that there are two or three errors per human genome (3 x 109 base pairs) per cell division, an acceptable rate. Inborn errors in the mismatch repair system are known in humans; they produce Hereditary Non-Polyposis Colorectal Cancer, or HNPCC, also known as Lynch Syndrome, one of the hereditary cancer predispositions.
As it happens, even if DNA replication occurs correctly, there is a risk that bases will change spontaneously by deamination. Cytosine can change to uracil, adenine can change to hypoxanthine, guanine can change to xanthine, and 5-methylcytosine can change to thymine. The latter two cases cause a change in base pairing, so upon replication, one of the two daughter strands will be fixed for the mutation.
Besides spontaneous mutations that cause base changes, there are many chemicals and other aspects of the environment that are mutagenic. We will not discuss in detail: 1) alkylating agents that modify bases so that upon replication, they base pair with the wrong base, 2) intercalating agents like proflavin that slip between bases of the double helix and cause base additions or deletions upon replication, 3) base analogs that are incorporated as one base, but cause replication errors by inappropiate base pairing.
We will briefly touch on mutations induced by UV irradiation. UV irradiation produces a very specific kind of mutation by creating thymine dimers. Two thymine bases that are adjacent on the same strand can become crosslinked by ultraviolet light, as shown in cartoon form below.
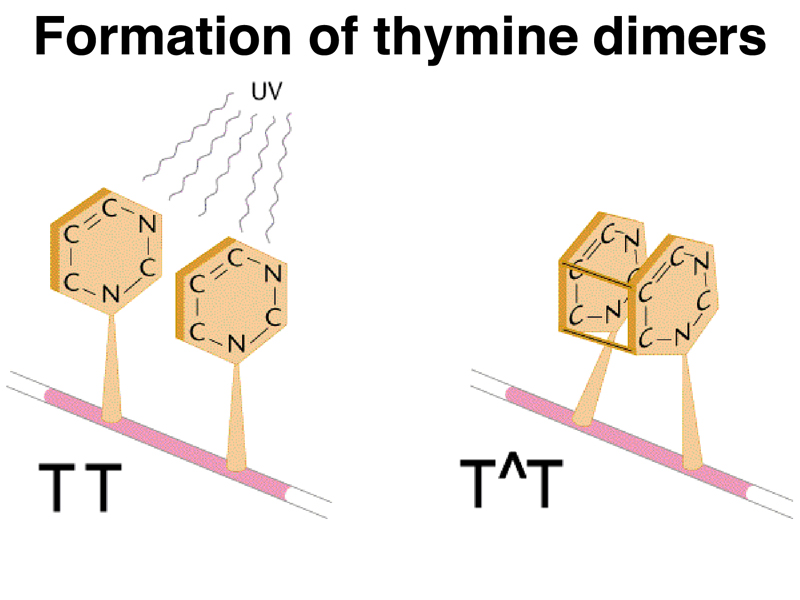
There is an excision repair system that removes thymine dimers and related lesions as shown below. First, a glycosidase removes the base from the deoxyribose backbone to generate an abasic site. This is repaired by either "long patch" (8-10 bases) or "short patch" (1-2 bases) repair systems. There are heritable variants in four genes that are part of the excision repair system in humans. All four genes produce a similar phenotype called xeroderma pigmentosum. One of these genes, XRCC1, encodes a protein partner to DNA ligase as shown in the diagram below.
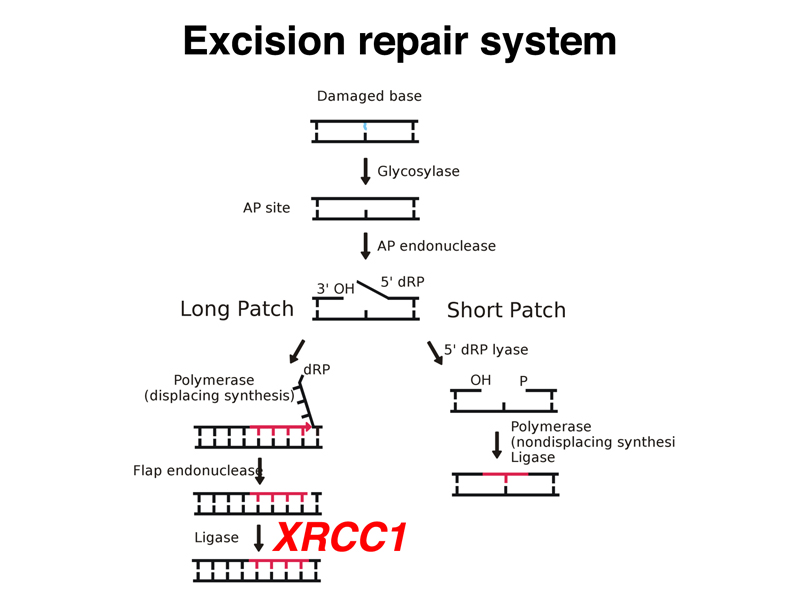
Xeroderma pigmentosum is characterized by extreme photosensitivity to UV light. Exposure causes patients to develop multiple basal cell carcinomas in exposed skin. We saw some gruesome slides, as well as a slide of a young patient wearing UV protective gear (a UV-protective clear face shield and a shroud of UV-opaque material). There is no other current treatment.
We reviewed the different types of point mutations in an overview, presented below, before looking more closely at specific examples.
 |
Silent. A silent mutation in a coding region is the substitution of a synonymous codon for the original codon. Because the amino acid encoded is the same, such mutations are not likely to have any phenotypic effect. Substitution of one stop codon for another is also generally a silent mutation. We can also think of base substitutions in noncoding regions (98% of the human genome) as silent mutations, as the overwhelming majority of such mutations will not have any phenotypic effect. |
 |
Missense. A missense mutation is the substitution of a nonsynonymous codon for the original codon. The amino acid replacement might be a conservative substitution (like isoleucine for valine) where the two amino acids are chemically similar, or a nonconservative substitution (like cysteine for tryptophan as shown here) where the two amino acids are very different chemically. |
 |
Nonsense. A nonsense mutation is the substitution of a stop (nonsense) codon for the original codon. Nonsense mutations cause the termination of protein synthesis, and if they occur anywhere but very near the 3' end of the gene, are likely to cause a complete loss of gene function. |
 |
Frameshift. In a frameshift mutation, a base is inserted or deleted, altering the codon in which the insertion or deletion took place, but also changing the reading frame so that all codons downstream are read out of frame. This typically produces a string of amino acid substitutions before a stop codon is reached. Stop codons are fairly frequent in coding sequences read out of frame. Frameshift mutations can be caused by the insertion or deletion of any number of bases that is not an integral multiple of three. |
 |
Splice Site. Mutations of splice sites (unfortuantely not covered in the textbook) account for about half of all disease-causing mutations in humans. Recall that a splice donor is a GT pair at the 5' end of an intron that is part of a consensus sequence, and that a splice acceptor is an AG base pair at the 3' end of an intron that is part of a consensus sequence. In the example shown here, the splice donor site mutates via a single base substitution such that it can no longer be a splice donor. The consequence in this case is shown in the image below. |
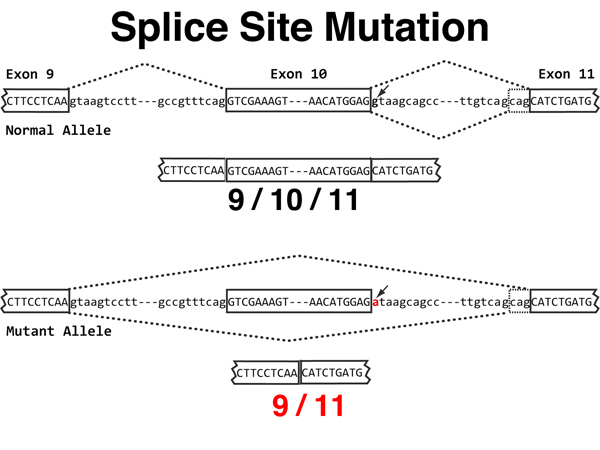 |
Splice Site. In the original sequence at the top of the image, exon 9 joins exon
10 as the first intron shown is spliced out. There is an alternative splice acceptor for the
second intron, producing two slightly different splices of exon 10 to exon 11.
The single splice donor for the second intron is mutated in the sequence at the bottom
of the image.
Loss of the splice donor for the second intron causes the splicing machinery to use the prior splice donor. This causes exon 9 to be spliced to exon 11, entirely omitting exon 10. The effect on the synthesis of the protein is the loss of all of exon 10. In addition, it is possible that the novel splice junction will cause a frameshift immediately downsteam of the new splice. |
Silent Mutations. There isn't much more to say about silent mutations at this time, although they are very useful as markers in human genomics because they are selectively neutral.
Missense Mutations. As an interesting example of the effects of missense mutations, we looked at alleles of FBN1 that cause Marfan Syndrome, discussed in a prior lecture. The protein fibrillin is synthesized inside cells (as are all proteins), then exported for assembly into elastic fibers and microfibril bundles that are part of connective tissue. All of the clinically significant alleles of FBN1 that cause Marfan Syndrome are dominant. We can understand why this is so if we consider the effects of assembling a variant protein into a complex macromolecular assembly. If half of the protein in the assembly is defective, the whole structure is defective.
Fibrillin is extensively crosslinked in elastic fibers and microfibril bundles. The crosslinking is a postranslational modification that connects one protein molecule to another. It is useful to think about this when we look at the first eight missense alleles listed in the OMIM entry:
FBN1-R1137P (ARG --> PRO)
FBN1-C2307S (CYS --> SER)
FBN1-C1249S (CYS --> SER)
FBN1-C1663R (CYS --> ARG)
FBN1-C2221S (CYS --> SER)
FBN1-N2144S (ASN --> SER)
FBN1-N548I (ASN --> ILE)
FBN1-D723A (ASP --> ALA)
When I first saw this list, I was struck by the number of missense alleles causing the loss of a cysteine (half of these eight). Reading the summary of the research in the OMIM entry, it is clear that the formation of disulfide bonds between fibrillin molecules is an important aspect of the assembly of elastic fibers and microfibril bundles. This explains why a simple missense allele of a gene encoding a large protein can produce a dominant phenotype.
Missense alleles of other genes, such as those encoding enzymes, can have a profound effect on protein folding or enzyme activity, but such mutations are typically recessive. A large number of missense alleles have no phenotypic effect, especially if the amino acid substitution is conservative.
Nonsense Mutations. Nonsense mutations typically produce alleles with a complete loss of gene function. As an example, we selected a nonsense allele of PAH, the gene encoding phenylalanine hydroxylase. Mutations in this gene are responsible for phenylketonuria (PKU), which we discussed in a prior lecture.
The PAH-R111X allele (X stands for a termination codon in this nomenclature, PAH-R111* is also used) accounts for about 10% of the clinically significant alleles of PAH in the Chinese population. The sequence of the normal PAH protein and the sequence of the protein produced by the PAH-R111X allele are shown below. The red X in the sequence of the PAH-R111X allele is the stop codon.

Students in the class agreed that it was very unlikely that there was any enzyme activity in the protein encoded by the PAH-R111X allele.
Comparison of the sequences of the two proteins shows that the new stop codon was formerly an arginine (R) codon. There are six arginine codons (CGU, CGC, CGA, CGG, AGA, and AGG) and three stop codons (UAA, UAG, and UGA). Examination of these codons suggests that the mutation is CGA --> UGA or AGA --> UGA, as these are the only single-base substitutions that can generate a stop codon. Interested students can look up the answer to see if this is correct.
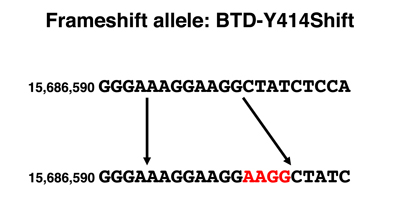
Frameshift Mutations. Frameshift mutations typically produce alleles with a complete loss of gene function. As an example, we selected BTD-Y414Shift, a private allele first discovered in my own genome (I am heterozygous, so I'm OK). The BTD gene encodes biotinidase, an enzyme necessary for recycling the vitamin biotin. Biotin is covalently attached to three different carboxylases as a cofactor. When these enzymes are recycled into amino acids, the enzyme biotinidase cleaves the biotin from the lysine to which it is covalently attached for reuse. Mutations in biotinidase are recessive, as is typical for most enzymes. People homozygous for loss-of-function variants of BTD require supplemental biotin in excess of the normal dietary requirement. This is one of the biochemical conditions that is identified by newborn screening (first instituted for PKU). You can read more about Biotinidase deficiency at OMIM.
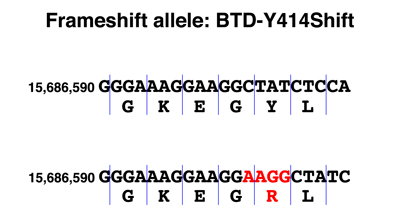
The genomic DNA sequence showing the alteration in the BTD-Y414Shift allele is shown below, together with a drawing comparing the DNA sequence of the normal and frameshift alleles parsed into codons. The sequence of the normal protein and the protein encoded by the BTD-Y414Shift allele are shown below. The amino acids from the position of the frameshift are shown in red.
 |
 |
 | |
We see from the DNA sequence that the BTD-Y414Shift allele has a four-base insertion. Examination of the sequence around the insertion shows that the sequence AAGG occurs twice in the normal allele and three times in the BTD-Y414Shift allele. Mutations that cause repeat expansion result from errors in replication or from recombination between sequences that are out of register and are known in many other genes.
When we parse the sequence into codons, we see that the first change is the substitution of an arginine (R) for a tyrosine (Y). Although the second amino acid is still leucine (L), this is just a coincidence. From the arginine codon on, the sequence is read out of frame, as shown in the protein sequence. A string of incorrect amino acids occurs before a stop codon is reached. Because the segment of the protein after the frameshift has at least five amino acids at which missense alleles cause a loss of gene function, we are very confident that this allele is a complete loss of function.
Splice site mutations. Splice site mutations can eliminate an existing donor or acceptor site, which will cause an exon to be skipped and possibly result in a frameshift. They can also "awaken" a cryptic site that is close to the consensus but lacks the critical 5' GT or 3' AG. In this case, a new splice will be made that is likely to have a severe effect on protein function. Up to half of the disease-causing alleles identified in humans are splice site mutations.
Other mutations. We mentioned in passing a few other kinds of point mutations:
Now that we have surveyed how mutations can arise and the different kinds of point mutations, it is useful to survey variation in humans to see what we have learned from our study of the human genome. Human variants can be classified into types, some of which are point mutations.
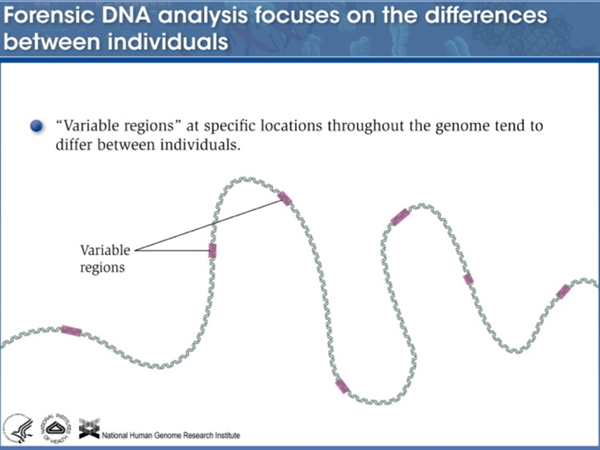
Single Nucleotide Polymorphisms (SNPs). Single nucleotide polymorphisms (SNPs) are sites of variation in the genome that exhibit single-base differences in the population. Where the reference sequence of the human genome has a T, there may be alleles with an A, C, or G at that position in the population. There is a SNP about every 1000 base pairs in the human genome. Any two individuals differ at about 6,000,000 SNPs from each other. Most SNPs are selectively neutral and represent ancient variation in our species. SNPs can be in noncoding sequence (as are the vast majority) or in coding sequence, where they may either be synonymous substitutions or other alleles that are tolerated. Because of the way that SNPs are identified (survey sequencing of individuals), some mutations that are under negative selection may be identified as SNPs that have very low allele frequencies in the population.
Copy Number Variants. Copy number variants are insertions or deletions of DNA anywhere from tens or hundreds of bases up to large duplications or deletions that are visible cytologically. While these are a significant component of human genetic variation, copy number variants above a certain size are not easily detected by current sequencing technology.
We discussed human copy number variants that are of special interest: short tandem repeat (STR) loci, also known as variable number of tandem repeat (VNTR) loci, that are used in the forensic analysis of human DNA. The slides from NHGRI summarizing forensic analysis of STR loci are presented below.
 |
The general idea here is to match DNA from a crime scene to DNA from a subject. There is usually not very much DNA at a crime scene, so we need to use a technique that can analyze very small amounts of DNA. The consequences of making mistakes in identification are serious, so we would like to use a technique that is very accurate. |
 |
We leave cells from our skin, hair, saliva, and other sources everywhere we go. You have probably seen some movies or television shows where DNA is recovered from sources like this. |
 |
The human genome has a large number of locations of short sequences that are repeated a number of times, flanked by unique DNA. The number of repeats at any particular locus is highly variable. These are typically not in coding regions. |
 |
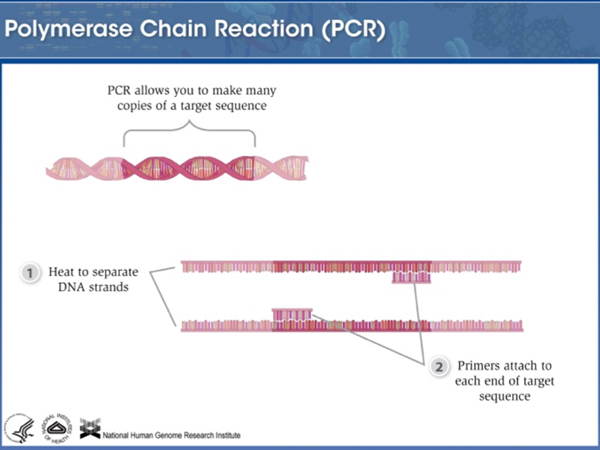
Here is an example of a short tandem repeat of four bases. |
 |
Different alleles of this particular STR are very different. Most individuals are heterozygous, with each of their alleles carrying a different number of copies of the repeat. |
 |
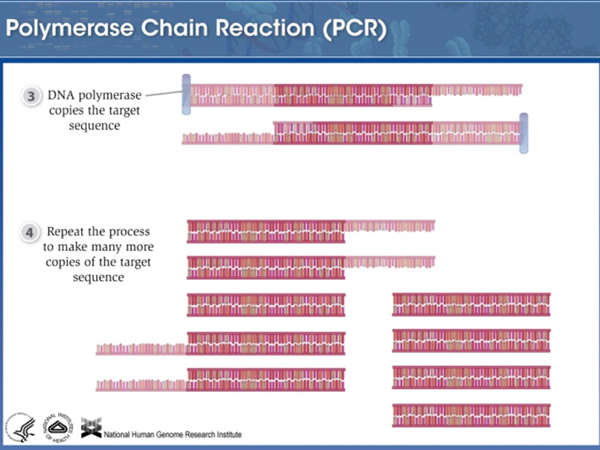
We can use Polymerase Chain Reaction (PCR) to amplify a particular STR using primers to the flanking unique sequence. |
 |
Thirty cycles of PCR is enough to produce a billion copies of the target sequence from a single molecule. |
 |
Because the primers hybridize to flanking unique sequence, alleles with different numbers of repeats can be amplified equally well. |
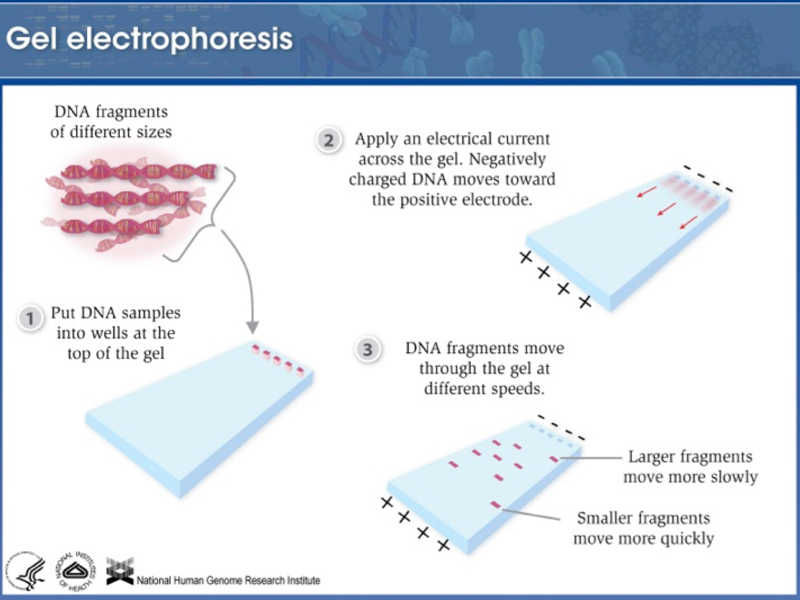 |
Amplified DNA is loaded onto an agarose gel and subjected to electrophoresis. DNA is negatively charged because of the sugar-phosphate backbone, so it runs to the cathode in an electric field. In an agarose gel, small molecules run faster than large molecules, so amplified fragments are sorted by size. |
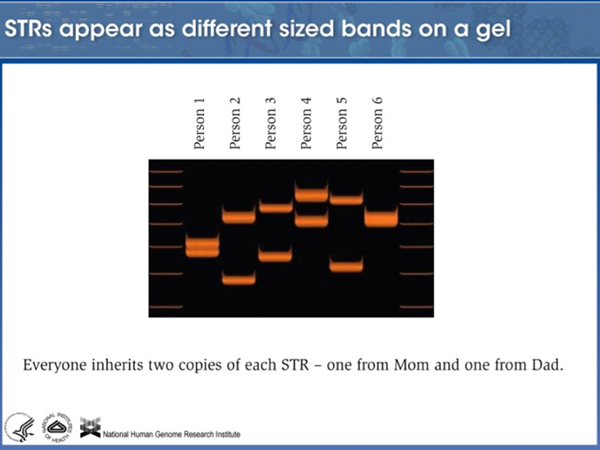 |
Here is a photo of a gel of an amplified STR locus from six individuals. Notice that every individual has two different bands of different size, showing that this STR is highly polymorphic. It looks like these six individuals carry at least ten different alleles (the maximum number would be 12). |
 |
There are many STR loci; forensic analysts at the FBI have settled on 14 of them that behave well. |
 |
There is only one chance in a billion that you will match someone unrelated to you. You will partially match your relatives, so a "DNA sweep" of a neighborhood might pick up an inexact match that would lead investigators to test relatives of that individual. Needless to say, a DNA sweep of a neighborhood is a controversial police tactic, although it has been used. |
Mutations. Even though SNPs and STR variants are also mutations, sometimes in human genetics the term is used in a restrictive sense to apply to variants in coding regions. How many mutations does a average person carry? You might be surprised to learn that the number is very large.
To explore this subject, I showed two slides with the summary of the analysis of my exome (the coding regions of my genome). Only about 2% of the human genome is coding sequence. Exome sequencing allows only that part of the genome likely to have the largest impact on inherited conditions to be analyzed. The first image, shown below, sorts variants into bins based on their expected impact.
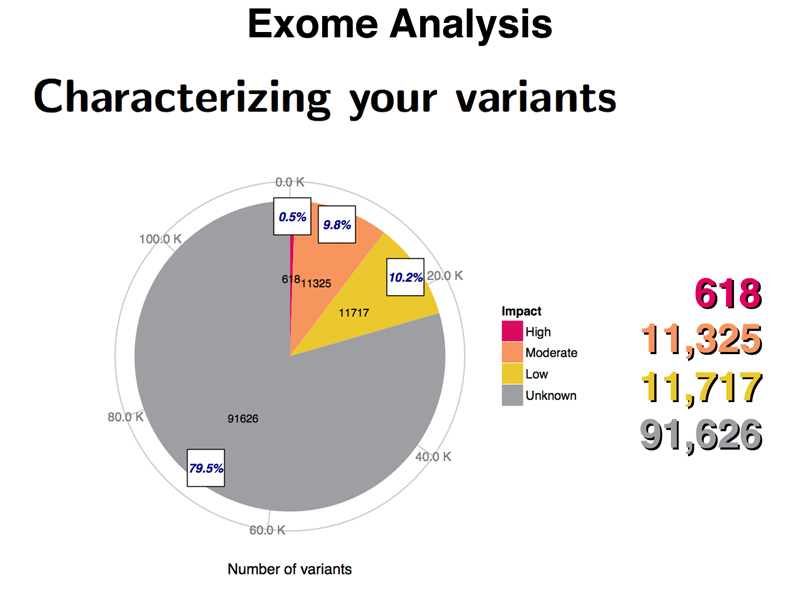
The exome analysis identified 618 variants likely to have a high impact in my exome, based on the kind of mutation. These include frameshifts, splice site variants, nonsense mutations, start-loss mutations, and stop-loss mutations. While I will be homozygous (or compound heterozygous) for a few of these, for most of these variants, we expect the other copy of the gene to be functional.
There are over 11,000 variants of moderate impact. These include nonsynonymous substitutions as well as insertion or deletion of whole codons (insertions or deletions that don't cause frameshifts). Some of these missense alleles are known to be selectively neutral.
There are over 11,000 variants of low impact. These are synonymous substitutions, synonymous stop codon substitutions, and start-gain mutations.
There are over 91,000 variants of unknown impact. These are unlikely to affect gene function, and include variation in the 3' UTR, 5' UTR, or portions of the introns that were sequenced during exome sequencing.
There is no particular reason to think that there is anything unusual about my genome, so the results obtained by analyzing the exome of any student in the class would be similar.
The other way to evaluate my variants is by their frequency in the population. Common variants that have an allele frequency of over 5% in the population are unlikely to be pathogenic. As shown in the figure below, 83% of my variants (95,660 variants) are of this type. This probably includes all of the 91,000 variants of unknown impact.
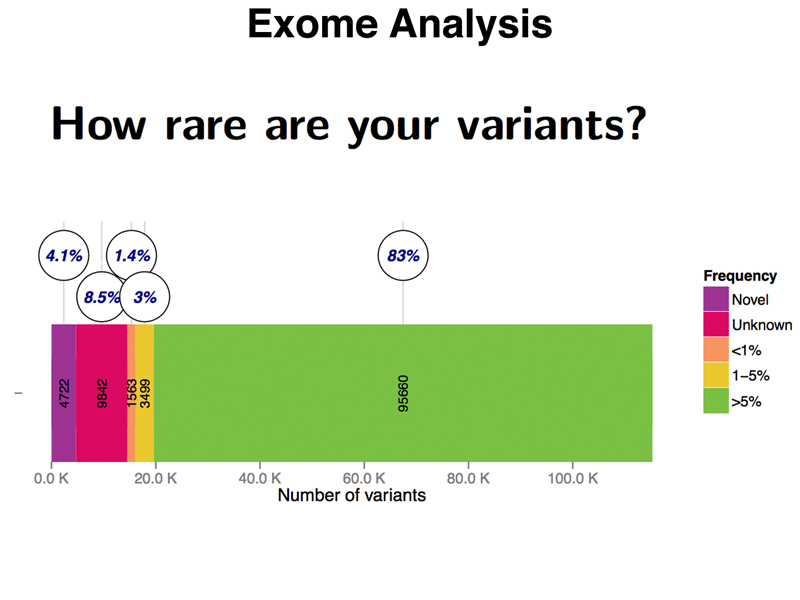
There are 3,499 variants that are uncommon (1-5% allele frequency), 1,563 variants that are rare (less than 1% allele frequency), and 9,842 variants of unknown frequency, meaning that they have been seen in other people, but that there is not yet a good estimate of their allele frequency. Surprisingly, I have 4,722 novel variants that have never been seen in other people, despite the thousands of people whose genomes are represented in public databases. Until we have sequenced hundreds of thousands, or even millions of people, sequencing individual exomes from other people will produce similar results.
Chromosome Rearrangements. Chromosome rearrangements include duplications, deletions, inversions, translocations, and transpositions. We are very fond of the subject of chromosome rearrangements, but are not able to present it in detail in this course.
We have talked about a number of inherited diseases in people. Most of the ones that we have talked about have devastating consequences. This tends to creat the impression that mutations are all terrible things that have enormous consequences for our health. In order to counter this impression, we like to present "superpower" mutations that have positive consequences under some circumstances. Here are three of my superpowers.
Norovirus/rotovirus resistance. The "stomach flu" that breaks out on cruise ships is not influenza. It is caused by norovirus. Norovirus, rotovirus, and Helicobacter pylori, the bacterium that causes stomach ulcers, all use a particular cell surface antigen to bind to cells in the gastrointestinal tract. They use the ABO blood group antigens, which are also present on the cells lining the GI tract in most people. As I am AB+, this might be a source of concern for me, except that I am homozygous for FUT2-W154*, a nonsense allele of the fucosyltransferase that adds these carbohydrates to a cell surface protein in the GI tract. As a consequence, I am resistant to norovirus, rotovirus, and Helicobacter pylori.
Reduced serum triglycerides and LDL. I am heterozygous for ANGPTL4-E40K, which reduces my serum triglyceride level (and LDL) by 15%. I have always had good numbers when screened for triglycerides and LDL, and previously thought that it was the healthy choices in my diet, but as it happens, I have had some help from this minor superpower that protects against heart disease.
Resistance to starvation and the Black Death. I am a compound heterozygote for two mutations that increase my risk of developing hemochromatosis, a disorder associated with the accumulation of excess iron from the diet. This doesn't sound like much of a superpower at first, but my genotype of HFE-C282Y/HFE-H63D makes me resistant to the Black Death and somewhat more resistant to iron deficiency during starvation. Both of my parents survived starvation during World War II, and my remote ancestors survived many outbreaks of the Black Death in Europe. The same defect that keeps me from properly regulating iron uptake from my diet is responsible for a low-iron environment in my macrophages. Iron transport in the gut and in macrophages is regulated by the destruction of the iron transport system by proteolysis when serum iron levels hit a certain level. Macrophages tend to be iron-rich, because they consume senescent red blood cells. In normal people with optimal serum iron levels, macrophages accumulate iron. People like me who have a defect in regulating the destruction of the iron transport system export iron from macrophages constitutively.
When a person is infected with Yersina pestis from a flea bite, macrophages take up the bacterium for transport to the lymph nodes. It turns out that the rate-limiting nutrient for Yersina pestis is iron. Normal people therefore transport a raging bacterial infection to their lymph nodes, which swell to form the bubos of bubonic plague. Iron-deficient macrophages in people like myself do not give Yersina pestis the boost that it needs to conduct a successful infection.
Selection for variants of HFE like the ones that I carry make hemochromatosis the most common genetic disorder in people of European descent. The disease tends to develop late in life. I am staving it off through changes to my diet (not much red meat) and frequent blood donations to reduce my iron load.
I don't have these variants, but they are fun to talk about.
Super muscles. People homozygous for mutations in MSTN (myostatin) have "double muscles" like the famous Belgian cattle and comparable dogs and mice. Only a few such people have been identified so far. I showed a photo of a seven-month-old child who was totally ripped.
Superstong bones. People with gain-of-function variants of LRP5 have superstrong bones. They often discover their superpower during surgery when surgical saws and drills break on their bones. Obviously, they are resistant to fractures. The downside is that they are poor swimmers because they are negatively bouyant.
HIV resistance. People homozygous for loss-of-function alleles of CCR5 cannot be infected by HIV. HIV uses this nonessential protein to enter T cells. This variant was discovered among sex workers in Africa who remained HIV negative after exposure to multiple partners infected with HIV. In a famous case, a man who had both HIV infection and leukemia was cured of both conditions by a bone marrow transplant from a donor lacking functional CCR5.